Preimplantasyon Genetik Tanı - Tek Gen Hastalıkları
Ailesel genetik geçiş gösteren tek gen ile aktarılan hastalığın bulunduğu çiftlerin embriyolarında PGT-M işlemi ile genetik hastalığa yol açan mutasyonu taşımayan sağlıklı embriyoların seçimi yapılabilir. Bu test işleminde ilk önce hastalığa sebep olan mutasyonu taşıyan gen bölgesinin detaylı haritası çıkartılarak, mutasyon bölgesi ve çevresi tanımlanır. Testin ilk basamağında IVF tedavisi ile elde edilen embriyolardan alınan biyopsi örneklerinin DNA çoğaltma işlemi gerçekleştirilir. Ardından mutasyon analizi ile mutasyonu içermeyen tek gen açısından sağlıklı embriyolar seçilir. Daha sonra sağlıklı embriyoların kromozom sayılarının tespiti için PGT-A işlemi gerçekleştirilebilir. Böylece hem tek gen açısından sağlıklı ve hem de kromozom sayısı açısından doğru sayıda kromozom taşıyan embriyolar seçilebilir.
Genetik bilimindeki son yıllardaki gelişmeler; tüp bebek yöntemiyle geliştirilen embriyolarda genetik incelemeler yapılmasına imkân tanımaktadır. Gebelik öncesi genetik tanı adı da verilen bu işlem; yumurta ve sperm hücrelerinin laboratuvar ortamında döllenmesi sonucunda gelişen embriyolardan alınan hücre(lerde) gerçekleştirilmektedir. Alınan bu hücrelerde özel yöntemler kullanılmakta ve doğacak bebekteki sayısal ve yapısal kromozom bozuklukları ile tek gen hastalıklarının (Akdeniz anemisi, Orak hücreli anemisi, Kistik fibrozis gibi) tanısı yapılabilmektedir. Böylece sağlıklı embriyoların anne adayına transferi ile sağlıklı bebeklerin doğması sağlanmaktadır.
Dünyada ilk PGT bebeği 2000 yılının Ekim ayında Amerika Birleşik Devletlerinde dünyaya gelmiştir. Bu gebelikte PGT yöntemi kullanılarak, yaklaşık 15 embriyo arasından tek sağlıklı olanı seçilip transfer edilmiştir.
PGT-M’de Amaç
Bireylerin; taşıdıkları kalıtsal hastalığı değişik oranlarda çocuklarına aktarma riskleri nedeniyle genetik hastalıkların bireylerde ve embriyolarda belirlenmesi çiftlerin sağlıklı çocuk sahibi olabilmesi için önemlidir. Günümüzde; farklı teknikler kullanılarak, birçok kalıtsal hastalığın tüp bebek aşamasında embriyolar ana rahmine konmadan önce tanımlanması mümkün hale gelmiştir. Preimplantasyon Genetik Tanının amacı, genetik hastalıkların gebelik öncesi dönemde yani henüz embriyo aşamasında tanımlanmasıdır.
PGT-M Hangi Durumlarda Önerilmektedir?
PGT-M, genetik bir hastalığı olan veya kalıtsal bir hastalık için taşıyıcılık saptanmış ve sağlıklı çocuk sahibi olmak isteyen çiftlere önerilmektedir.
Özellikle, tek gen hastalıkları veya kromozom bozukluğu saptanmış çiftlerin çocuklarında hastalık riski yüksek olduğu için günümüzde sıkça uygulanmaktadır.
Benzer olarak, ailesel kansere yatkınlık sendromlarına neden olan genetik değişikliklerin saptandığı çiftlerde de kullanılabilmektedir.
PGT-M SÜRECİ
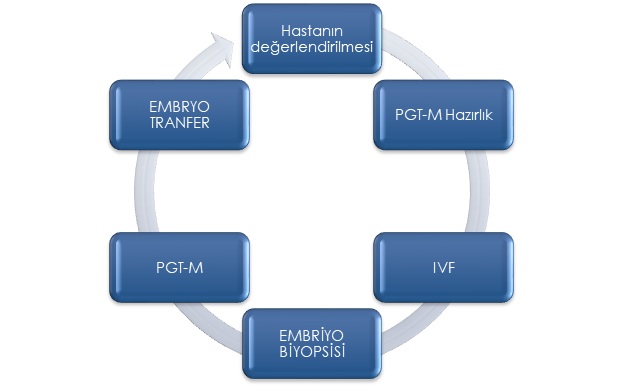
Tek Gen Hastalıklarında Preimplantasyon Genetik Tanı
Bugüne kadar tek gen hastalıklarıyla ilişkilendirilmiş toplamda 5.000’in üzerinde (monojenik) mutasyon tanımlanmıştır.
Vücudumuzdaki tüm hücrelerin özel bir genetik şifre içerdiğini ve bu şifrenin her bireyde birbirinden farklı özellikler taşıdığını bilmekteyiz.
Hücrelerimizin tüm görevleri bu genetik şifreler doğrultusunda planlanmaktadır.
Genetik şifredeki küçük değişiklikler bazen telafisi olanaksız eksikliklere veya hasarlara yol açarak genetik hastalıkların oluşmasına sebep olmaktadır.
Son yıllara kadar bu tip hastalıkların tanısı sadece klinik olarak tanımlanabilmekteydi. Genetik bilimindeki gelişmeler ve genetik şifrelerin çözülmesine yönelik çalışmalar sayesinde bu hastalıkların tanısı gen düzeyinde konulabilmektedir.
PGT-M ile Hangi Tek Gen Hastalıklara Tanı koyulabilmektedir?
İstanbul Memorial Hastanesi Genetik Tanı Merkezi’nde mutasyonu belirlenen tüm genetik hastalıklar için Preimplantasyon Genetik Tanı uygulaması yapılabilmektedir. Bugüne kadar Memorial Hastanesi Tüp Bebek Merkezi’nde 441 ailede 630 PGT siklusunda 138 farklı tek gen hastalığı için PGT uygulanmıştır. Ayrıca 314 ailenin 628 siklusunda HLA tiplemesi yapılmıştır. Merkezimiz bu alanda farklı genetik hastalıklara uygulanan PGT işlemleri açısından dünyanın en önde gelen merkezlerinden biridir.
PGT-M Ne İçindir?
Belirli bir genetik hastalığa sahip bireylerin hamilelikten önce etkilenmiş bir çocuğa sahip olmamak için in vitro fertilizasyon (IVF) yoluyla oluşturulan embriyoların test edilmesini ve daha sonra etkilenmeyen embriyoların transfer edilmesini sağlayan bir tanı yöntemidir.
PGT-M Kimlere Yapılır?
Siz ve eşiniz aynı genetik hastalık (otozomal resesif) için taşıyıcıysanız (örn. Kistik fibroz, Beta talasemi )
X'e bağlı bir hastalık taşıyıcıysanız (örneğin, Duchenne Musküler Distrofisi, Frajil-X)
Siz veya eşinizden biri genetik (otozomal dominant) hastalığa sahipseniz (örneğin Huntington hastalığı)
Siz veya eşiniz kalıtsal bir kanser sendromu ile ilişkili bir genetik değişime sahipseniz (ör. BRCA1 & 2)
HLA doku uyumlu nakil ihtiyacı olan bir çocuğunuz var ise.
Genetik hastalıklar bireyin DNA’sında meydana gelen değişimler (mutasyonlar) sonucu ortaya çıkan hastalıklardır. İnsandaki genetik hastalıkları üç ana grup altında incelemek mümkündür.
Kromozomal hastalıklar
Tek gen (Mendeliyen kalıtım ile aktarılan) hastalıkları
Mendeliyen kalıtım ile aktarılmayan hastalıklar
Kompleks kalıtım ile geçen
Mitokondri kalıtımı ile geçen
Tekrar dizi hastalıkları
Somatik hücre hastalıkları olarak sınıflandırabiliriz.
Tek Gen Hastalıkları
Tek gen hastalıkları, tek bir gendeki değişikliklerin hastalık sürecine dahil olduğu ve genellikle karakteristik kalıtım,
Otozomal dominant (baskın),
Otozomal resesif (çekinik)
X kromozomuna bağlı geçiş sergileyen
Mendel rahatsızlıklarıdır.
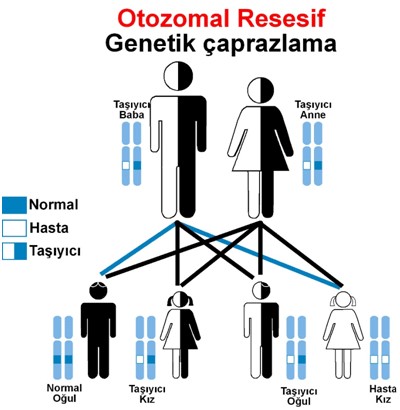
Şekil 1: Otozomal resesif kalıtımlı bir genetik hastalık taşıyıcı olan
çiftin çocukları için risk değerlendirilmesi.
Bu kalıtım en sık akraba evliliklerinde gözlenmektedir.
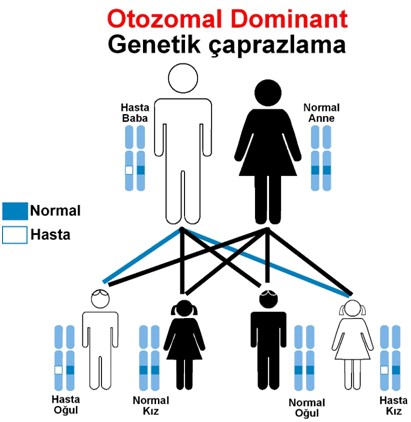
Şekil 2: Otozomal dominant kalıtımlı bir genetik hastalığın
kalıtımı ve çocukların risk durumu.
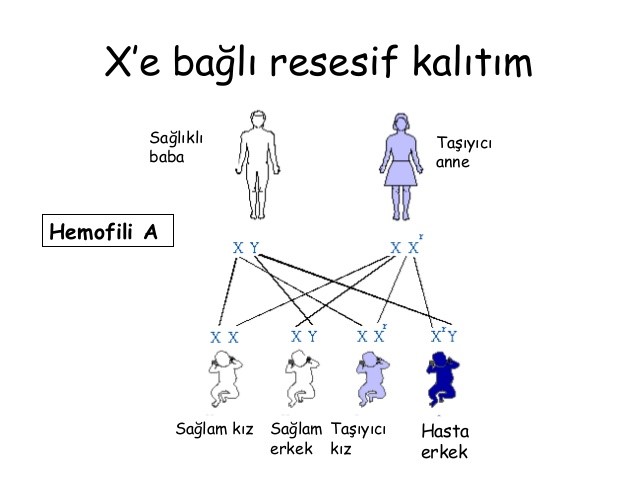
Şekil 3: X kromozomu ile taşınan kalıtsal hastalıkların riski.
Bu durumda anneden etkilenmiş X kromozomunu alan
erkek çocuklar hasta olmaktadır.
Sık Rastlanan Tek Gen Hastalıkları
Akdeniz Anemisi:
Akdeniz Anemisi (Beta-talasemi) ülkemizde en çok görülen kalıtsal hastalıklardan biridir. Hastalık geninin Türkiye'deki ortalama görülme sıklığı % 4-5 kadardır, fakat özellikle güney bölgelerde bu oran % 20'lere kadar çıkmaktadır. Beta-talasemi, otozomal resesif (genetik defektin hem anne hem babada taşındığı tipte) kalıtım gösterir. Akraba evliliklerinin yaygın olduğu bölgelerde hastalığın görülme sıklığı artmaktadır.
Beta-talasemi geni 11 numaralı kromozomun üzerinde yer almaktadır. Gen üzerinde oluşacak mutasyonlar beta-globin üretimini azaltır veya sentezlenmeyi durdurur. Hemoglobin üretimin azalması sonucu hastalarda mikrositik hipokromik anemi oluşur.
Beta-talasemi tanısı kan testleri yoluyla konulur. Periferik yaymada çekirdekli alyuvarların varlığı, hemoglobin elektroforezi sonucu hemoglobin A seviyesinin düşüklüğü hastalığın başta gelen bulgularıdır. Hastalığın tespit edilmesinden sonra mutasyonun tanımlanması moleküler tekniklerle yapılmaktadır. İstanbul Memorial Hastanesinde Beta-talasemi için moleküler tanı beta-globin geninin tümüyle dizilenmesiyle (Tüm Gen Analizi) konulmaktadır. Bu tanı yöntemi hem bireylerdeki mutasyonun tespit edilmesini hem de anne karnındaki fetüsten alınacak örnekle beta-talasemi mutasyonlarının tespitini sağlar. Gebeliğin 10. haftasından sonra Koryon villüs biyopsisi (CVS) veya 15. haftasından sonra yapılacak amniosentez yardımıyla olabilir.
Kistik Fibrozis:
Kistik Fibroz dünya genelinde sık rastlanan kalıtsal hastalıklardan bir tanesidir. Kistik fibroz, solunum ve sindirim sisteminin yüzeyini kaplayan hücrelerinde sodyum ve klor iyonlarının taşınmasını sağlayan proteini üreten CFTR genindeki mutasyonlar sonucu ortaya çıkar. Mutasyonlar sonucunda mukus tabakası daha yoğun bir hal alır ve akciğerlerin tıkanmasına ve enfeksiyon gelişmesine, pankreasın tıkanarak sindirim enzimlerinin bağırsağa ulaşımının engellenmesine neden olur.
Kistik fibroz transmembran iletim düzenleyicisi (CFTR) geni 7 nolu kromozomda yer alır. CFTR geninde yaklaşık 1400 tane mutasyon tespit edilmiştir. Mutasyonların oluşturacağı etkiler birbirlerinden farklıdır. Bazı mutasyonlar CFTR proteininin normalden küçük ya da yetersiz miktarda üretilmesine yol açarken, diğerleri proteinin regülasyonunu ya da hücre zarındaki lokalizasyonunu engellerler. Bazı mutasyonlar Kistik Fibroz hastalığına neden olurken CFTR geninde oluşan bazı mutasyonlar erkeklerde kısırlığa sebep olur. Bu mutasyonlar testislerden sperm taşıyan kanalların birinin (CUAVD: Congenital Unilateral Absence of Vas Deferens) veya her ikisinin de (CBAVD: Congenital Bilateral Absence of Vas Deferens) gelişmemesine neden olur. Testisler normal gelişmiş ve cinsel fonksiyonlar normal olmasına karşın vas deferens gelişmemiş olması nedeniyle erkek üreme sistemindeki spermler meniye taşınamaz.
Spinal Müsküler Atrofi (SMA)
Spinal müsküler atrofi (SMA), omurilikteki motor sinir hücrelerini etkileyerek hareket kabiliyetini kısıtlayan genetik geçişli bir kas hastalığıdır. Çocukları etkileyen en yaygın genetik hastalıklardan biridir. Dünya çapında her 6.000 ila 10.000 bebekten birinin SMA ile doğduğu tahmin edilmektedir. SMA hastalarının %95'inde SMN1 ekzon 7-8 delesyonu tespit edilmektedir.
Duchenne Müsküler Distrofisi (DMD)
Duchenne müsküler distrofisi (DMD), kas hücrelerindeki distrofin adı verilen bir proteini üreten distrofin genindeki mutasyonlar sonucu az üretilmesi veya üretilememesi sonucu oluşan ilerleyici kas dejenerasyonu ve zayıflığı ile karakterize genetik bir hastalıktır. DMD geni mutasyonlarının %60-65’i delesyon, %6’sı dublikasyondur. X’e bağlı resesif (XR) kalıtım göstermektedir, Erkekler hemizigot oldukları için, etkilenmişlerse mutlaka hastadırlar, kadınlar ise taşıyıcıdırlar.
Frajil-X Sendromu:
Frajil-X sendromu, kalıtsal zeka geriliğinin en sık nedeni olması yanında, aynı zamanda en sık görülen tek gen hastalığıdır. Bu hastalık daha çok erkek çocuklarında ortaya çıkmaktadır. Hastalık kızlarda ise taşıyıcı rol üstlenmektedir.
Frajil- X sendromu, X-kromozomu üzerinde bulunan FMR-1 adındaki gende oluşan ‘değişiklik’ (mutasyon) sonucunda ortaya çıkmaktadır.
FMR-1 geninin de , 5 ila 40 arasında yer alan ‘CGG’ tekrar sayısı Frajil-X hastalarında 200'den fazladır.
MERKEZİMİZDE ŞİMDİYE KADAR PGT - M YAPILAN HASTALIKLARIN LİSTESİ
|
HASTALIK ADI |
GEN ADI |
|---|---|
|
Mozaik Aneuploidi Sendromu 1 |
BUB1 |
|
Adenomatozis Polipozis Koli |
APC |
|
Adrenolökodistrofi |
ABCD1 |
|
Ailesel Akdeniz Ateşi |
MEFV |
|
Ailesel hemofagositik lenfohistiyositoz |
PRF1 |
|
Ailesel hipomagnezemi, hiperkalsiüri, nefrokalsinoz (FHHNC) |
CLDN16 |
|
Ailesel işitme kaybı |
GJB2 (Connexin 26) |
|
Akçaağaç Şurubu Hastalığı |
BCKDHA-BCKDHB |
|
Akondroplazi |
FGFR3 |
|
Alfa Talasemi |
HBA |
|
Alkuraya Kucinskas Sendromu |
KIAA1109 |
|
Alström Sendromu |
ALMS1 |
|
Argininosuksinat Liyaz Eksikliği |
ASL |
|
Artrogripoz-böbrek işlev bozukluğu-kolestaz (ARC) sendromu |
VIPAR |
|
Barter Sendromu |
BSND |
|
Batten Sendromu |
PPT1 |
|
Beta Talasemi / Orak Hücre Anemisi |
HBB |
|
Biotinidaz Eksikliği |
BTD |
|
Bloom Sendromu |
BLM |
|
BRCA-2 Ailesel Meme Kanseri |
BRCA2 |
|
Charcot-Marie-Tooth Tip1 |
CMT1-GJB1-PMP22-MFN2 |
|
Citrullinemi |
ASS |
|
Cockayne Sendromu |
ERCC6 |
|
Cowchock Sendromu |
AIFM1 |
|
Çok Uzun Zincirli Asil Koenzim A Dehidrojenaz Eksikliği |
ACADVL |
|
D-bifunctional Protein Eksikliği |
HSD17b4 |
|
Dermatosparaxis Ehlers-Danlos Sendromu (dEDS) |
ADAMTS2-SLC39A13 |
|
Diamond Blackfan Anemisi |
RPL5 |
|
Dilate Kardiyomiyopati |
MYBPC3 |
|
Distal Renal Tubular Acidosis |
ATP6V1B1 |
|
Distrofik Displazia |
DTDST |
|
Duane-Radial Ray Sendromu |
SALL4 |
|
Duchenne-Becher Müsküler Distrofi (DMD-BMD) |
DMD |
|
Embriyolarda HLA tiplemesi |
HLA |
|
Epidermolizis Bülloza |
KRT5-ITGB4-LAMA3-LAMB3-COL7A1 |
|
Facioscapulohumeral Müsküler Distrofi |
DUX4 |
|
Fankoni Anemisi |
FANCA |
|
Fenilketonüri Hastalığı |
PAH |
|
Fokal Segmental Glomerüloskleroz |
NPHS2 |
|
Fosfat Üridil Transferaz Eksikliği (Galaktozemi Tip 1- GALT Eksikliği) |
GALT1 |
|
Frajil-X Sendromu |
FMR1 |
|
Fraser Sendromu |
FRAS1 |
|
Gaucher Hastalığı |
GBA |
|
Gerstmann-Straussler-Scheinker (GSS) Sendromu |
PROSER1 |
|
Glikojen Depolama Hastalığı Tip 1(GSD I) |
G6PC |
|
Glukoz-6-fosfat Dehidrojenaz Eksikliği |
G6PD |
|
Gm1 Gangliosidozis |
GLB1 |
|
Griselli Sendromu |
MYO5A |
|
Hemofagositik Lenfohistositoz Tip 3 |
UNC13D |
|
Hemofili A |
F8 |
|
Hemofili B |
F9 |
|
Herediter Multiple ekzositoz |
EXT1-EXT2 |
|
Hiper IgE Sendromu |
DOCK8 |
|
Hiper IgM Sendromu |
CD40LG |
|
Hipofosfatazya |
ALPL |
|
Hipohidrotik ektodermal displazi sendromu |
EDA1 |
|
Hipomyelinizan Lökodistrofi |
POLR3A |
|
Holoprozensefali Tip 5 |
ZIC2 |
|
Homosistinüri |
MTHFR |
|
Huntington Hastalığı |
HTT |
|
Hipomyelinizasyon and Kongenital Katarakt |
FAM126A |
|
IVIC Sendromu |
SALL4 |
|
İlerleyici ailevi intrahepatik kolestaz (PFIC1/2) |
ABCB11 |
|
İnfantil Nöroaksonal Distrofi |
PLA2G6 |
|
İnfantil Serebellar Retina Dejenerasyonu |
ACO2 |
|
İnfantil Striatonigral Dejenerasyon |
NUP62 |
|
Jeune Sendromu |
WDR60 |
|
Joubert Sendromu |
AHİ1-TCTN2-CEP290-CC2D2A |
|
Karnitin Açil Transferaz Eksikliği |
SLC25A20 |
|
Kistik Fibrozis |
CFTR |
|
Koenzim Q10 Eksikliği |
CoQ10 |
|
Kombine Oksidatif Fosforilasyon Defekti |
AIFM1 |
|
Kongenital Adrenal Hyperplasia |
CYP21 |
|
Konjenital Faktör VII Eksikliği |
F7 |
|
Konjenital Glikosilasyon Tip 2B |
MOGS |
|
Konjenital Glikozilasyon Defekti Tip Ij |
DPAGT1 |
|
Konjenital Hidrosefali Tip 2 |
MPDZ |
|
Konjenital Katarakt |
CRYGD |
|
Konjenital Müsküler Distrofi |
LAMA2 |
|
Krabbe Hastalığı |
GALC |
|
Kseroderma Pigmentosum |
XPA |
|
Lafora Hastalığı |
EPM2B |
|
Leber Kongenital Amaurosiz |
RPE65 |
|
Lesch-Nyhan Sendromu |
HPRT1 |
|
Li-Fraumeni Sendromu |
TP53 |
|
Limb Girdle Musküler Distrofi |
LGMD |
|
Marfan Sendromu |
FBN1 |
|
Meckel Sendromu Tip 3 |
TMEM67 |
|
Metakromatik Lökodistrofi |
ARSA |
|
Metilmalonik Asidemi |
MMUT |
|
Mikrosefali, Büyüme Kısıtlaması ve Kardeş Kromatid Değişimi 2 |
TOP3A |
|
Miyotonik Distrofi |
DMPK1 |
|
Molibden Kofaktör eksikliği |
MOCS2 |
|
MPS Tip II/Hunter Sendromu |
IDS |
|
Mukolipidozis Tip 1 (Sialidosis Tip 1 ve Tip 2) (Nöraminidaz Eksikliği) |
NEU1 |
|
Mukolipidozis Tip 2 (I cell) Hastalığı |
GNPTAB |
|
Mukopolisakkaridoz Tip 3a |
SGSH |
|
Mukopolisakkaridoz Tip 4a |
GALNS |
|
Mukopolisakkaridoz Tip VI (Maroteaux-Lamy) |
ARSB |
|
Multiple Endokrin Neoplazi Tip1 |
MEN1 |
|
Multiple Schwannoma |
LZTR1 |
|
Multiple Sulfataz Eksikliği |
SUMF1 |
|
N-asetilglutamat sentetaz NAGS Eksikliği |
NAGS |
|
Neiman Pick Tip A-B-C1-D |
SMPD1-NPC1 |
|
Nemalin Myopati |
NEB |
|
Nonketotik Hiperglisemi |
AMT |
|
Norrie Hastalığı |
NDP |
|
Nörofibromatozis Tip 1 |
NF1 |
|
Nöronal Seroid Lipofussinozis Tip2 |
TPP1 |
|
Omenn Sendromu |
RAG1 |
|
Ornitin Transkarbamilaz Eksikliği |
OTC |
|
Osteogenesis İmperfecta Tip 5 |
IFITM5 |
|
Osteogenezis İmperfekta Tip 1 |
COL1A1-COL1A2 |
|
Osteoporozis Pseudoglomia |
LRP5 |
|
Otozomal Recessive Osteopetrozis |
TCIRG1-RANK1-TNFRSF11A |
|
Pelizaeus Merzbacher Sendromu |
PLP1 |
|
Persistan hiperplastik primer vitreus |
ATOH7 |
|
Pirüvat karboksilaz yetersizliği (hiperlaktatemi, hipoglisemi) |
PC |
|
Polikistik Böbrek Hastalığı 4 (PKD4) |
PKHD1 |
|
Polikistik Böbrek Hastalığı Tip 1 |
PKD1-PKD2 |
|
Pompe Hastalığı (Glikojen Depo Hastalığı Tip II) |
GAA |
|
Pontocerebellar hipoplazisi, Tip 9 |
AMPD2 |
|
Pontoserebellar Hipoplazi Tip 1A |
VRK1 |
|
Propionik Asidemi (PCCA) |
PCCA |
|
Proprotein Konvertaz 1/3 Eksikliği |
PCSK1 |
|
Purin Nukleosit Fosforilaz Eksikliği |
PNP |
|
Retinitis Pigmentosa / Stargart Sendromu |
PROM1 / PRPH2-ABCA4 |
|
Retinoblastoma |
RB1 |
|
Rett Sendromu |
MECP2 |
|
Rizomelik kondrodisplazi punktata |
AGPS |
|
Sandoff Sendromu |
HEXB |
|
Seckel Sendromu |
CEP152 |
|
Spastic Paraplegia |
ATL1-SPAST |
|
Spherocytosis Tip 1 |
ANK1 |
|
Spinal Müsküler Atrofi (SMA) |
SMN1 |
|
Spinocerebellar Ataxia Type 2 (SCA2) |
ATXN2 |
|
SRP Tip 6 (Kısa Kosta Polidaktili Sendromu Tip 6) |
NEK1 |
|
Tay Sachs Hastalığı |
HEXA-GM2AP1 |
|
Tirozinemi Tip 1 |
FAH |
|
TNF Reseptörü ile İlişkili Periyodik Ateş Sendromu (TRAPS) |
TNFRSF1A |
|
Torg Sendromu |
MMP2 |
|
Tüberoskleroz |
TSC1-TSC2 |
|
Usher Sendromu |
MYO7A |
|
Vici Sendromu |
EPG5 |
|
Von Hippel Lindau Hastalığı |
VHL |
|
Von Willebrand Hastalığı (VWD) |
VWF |
|
Waisman Sendromu |
RAB39B |
|
Walker Warburg Sendromu |
POMPT2 |
|
Wanishing White Matter (Kaybolan Beyaz Cevher) lökodistrofi |
EİF2B5 |
|
Wilson Hastalığı |
ATP7B |
|
X'e bağlı agammaglobulinemi |
BTK |
|
Zelweger Sendromu |
PEX1 - PEX5 - PEX6 - PEX12- PEX13 |

